Asbtract
The genus Aeromonas is a common gastrointestinal pathogen associated with human and animal infections. However, their evolutionary dynamics and genetic diversity are still fragmented due to the high level of cross-species similarity. Here we investigated the pan-genome of 29 Aeromonas species, as well as Aeromonas species in microbial communities, to clarify their evolutionary dynamics and genetic diversity, with special focus on virulence factors and horizontal gene transfer events. Our study revealed an open pan-genome of Aeromonas containing 10,144 gene families. Different selective pressures operated on the diverse functions, with the single-copy core genes and most accessory genes experiencing purifying selection. The significant congruence between core-genome and pan-genome trees revealed that core genes mainly affected evolutionary divergences of Aeromonas species. Gene gains and losses revealed a high level of genome plasticity, exhibited by hundreds of gene expansions and contractions, horizontal transferred genes and mobile genetic elements. The selective constraints shaped virulence gene pools of these Aeromonas strains, in which genes encoding hemolysin were ubiquitous. Among them, A. aquatica MX16A seemed to be more resistant as it harbored most resistance genes. Finally, the virulence factors of Aeromonas in microbial communities are quite dynamic in response to environment changes. For example, the virulence diversity of Aeromonas in microbial communities could reach levels that match some of the most virulent Aeromonas species (such as A. hydrophila) in penetrated-air and modified-air packaging. Our work shed light on genetic diversity, evolutionary history and functional features of Aeromonas, which would facilitate the detection and prevention of infections.
Carousel

Data
The whole-genome of single isolated sequenced Aeromonas
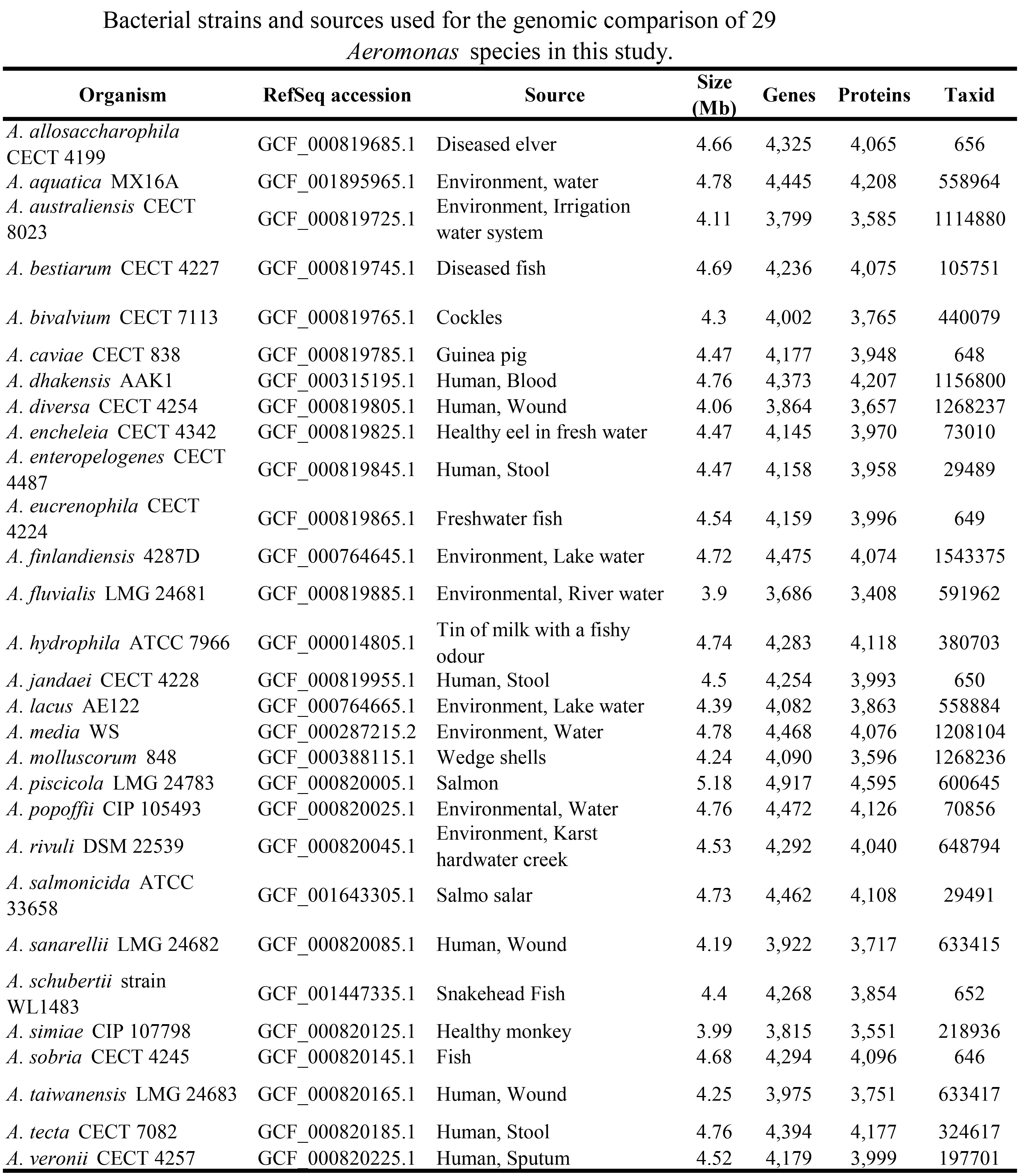
A dataset comprising 29 genomes (draft and complete) from the genome database of National Center for Biotechnology Information (NCBI) were obtained on Feb 14th, 2017. The genome scale study used the most complete sampling of the diversity of the Aeromonas species to date.
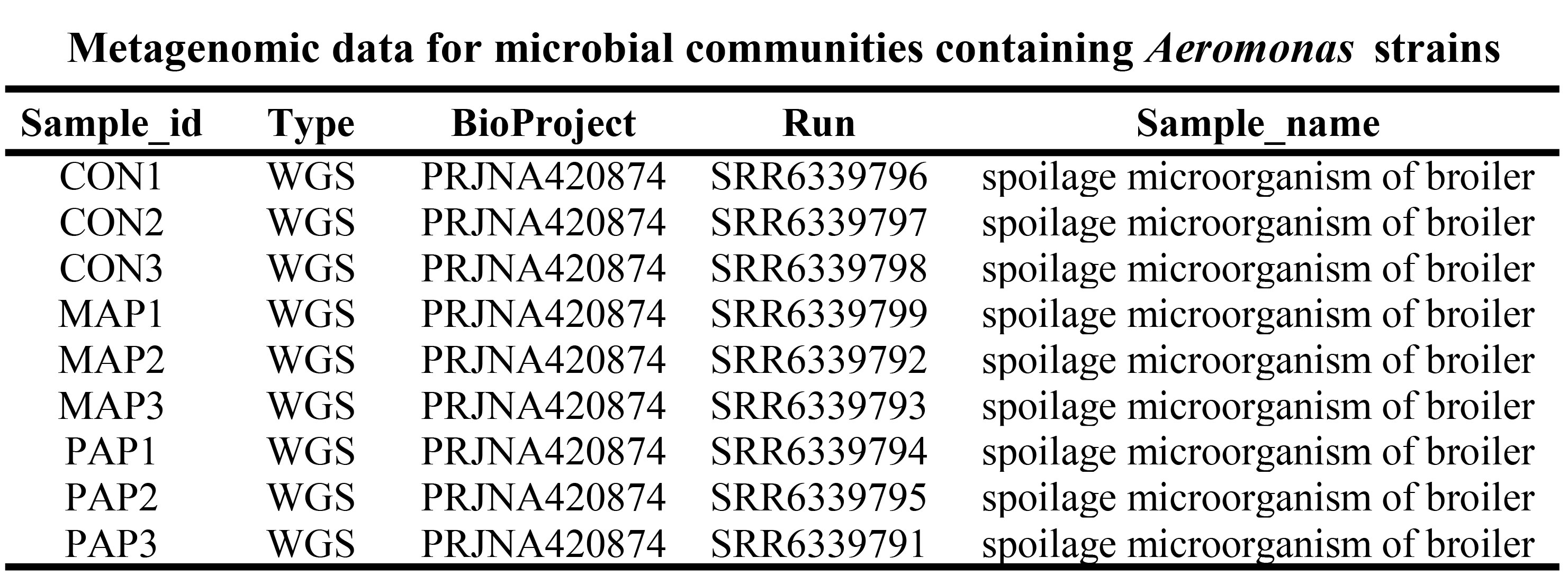
Metagenomic data for microbial communities containing Aeromonas strains
The metagenomes of chilled yellow-feathered broiler responding to modified-air packaging (MAP) and penetrated-air packaging (PAP) during storage and control samples with biosample identifier SAMN08123132 (BioProject accession: PRJNA420874; SRA: SRS2729591) were downloaded from NCBI.The Evolution and Pathogenicity of Aeromonas
Genetic diversity of Aeromonas
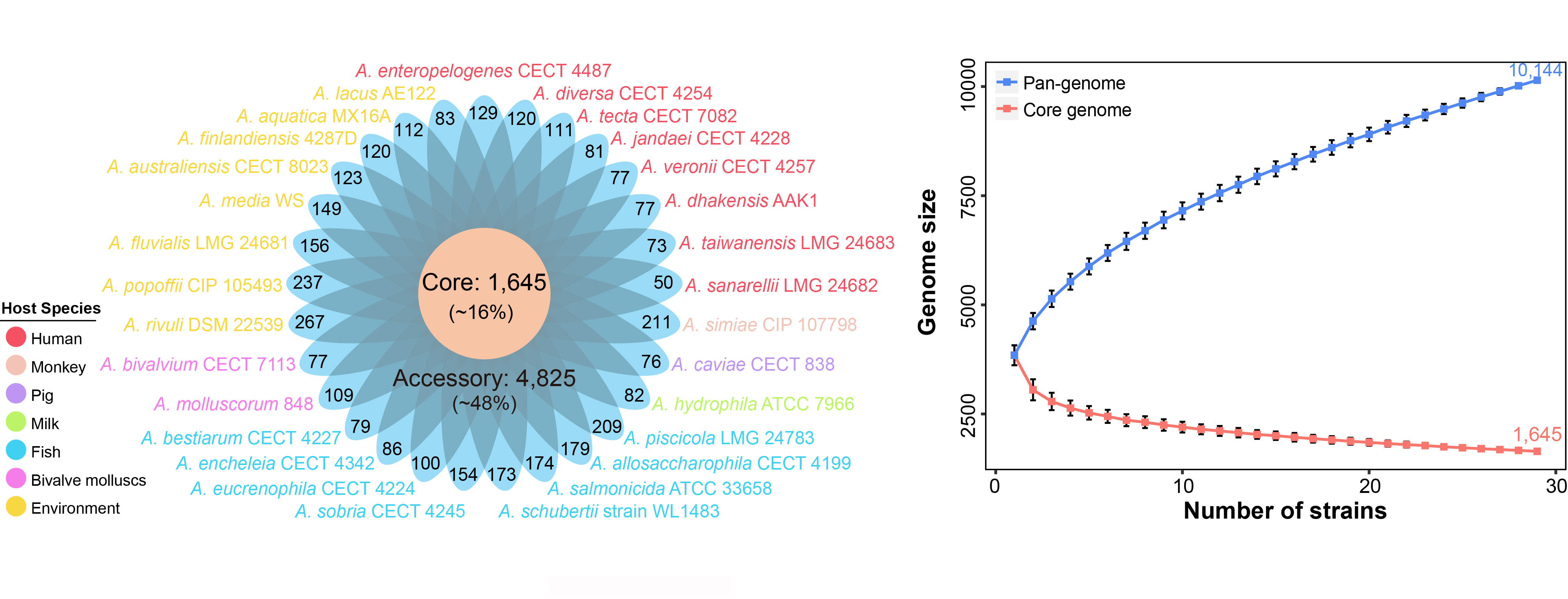
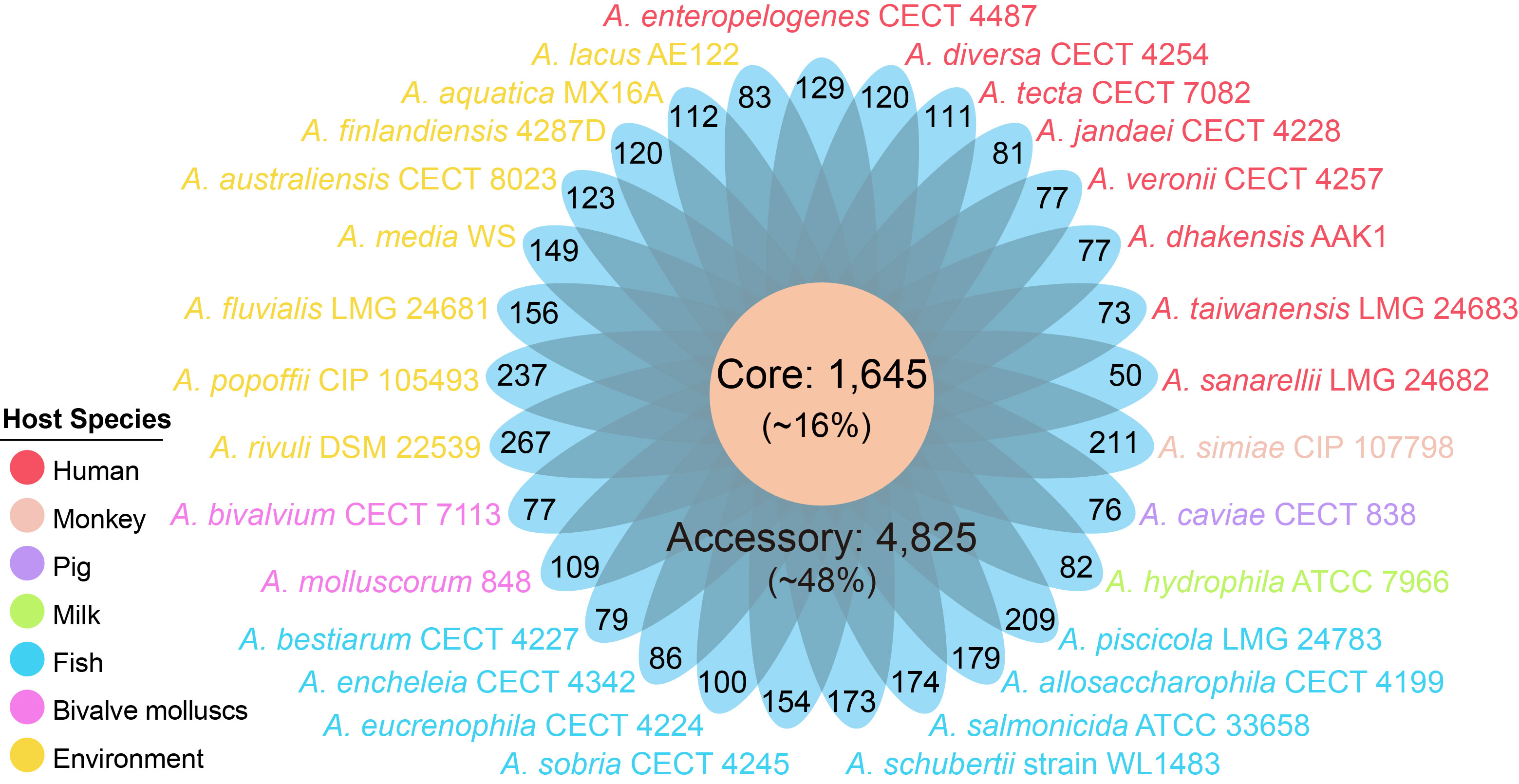
The 29 Aeromonas species contained 10,144 orthologous groups (defined as gene families), which were organized into core, accessory and unique genes. Among these gene families, 1,645 gene families (16%) shared by all strains constituted the core genome, 3,674 specific to a single strain constituted unique genome, and the remaining 4,825 present in more than one strain but not in all strains belonged to the Aeromonas accessory genome. In addition, the distribution of unique genes in Aeromonas were diverse, varying from 50 to 267, which further emphasized the heterogeneity of the genus, implying very high genome plasticity.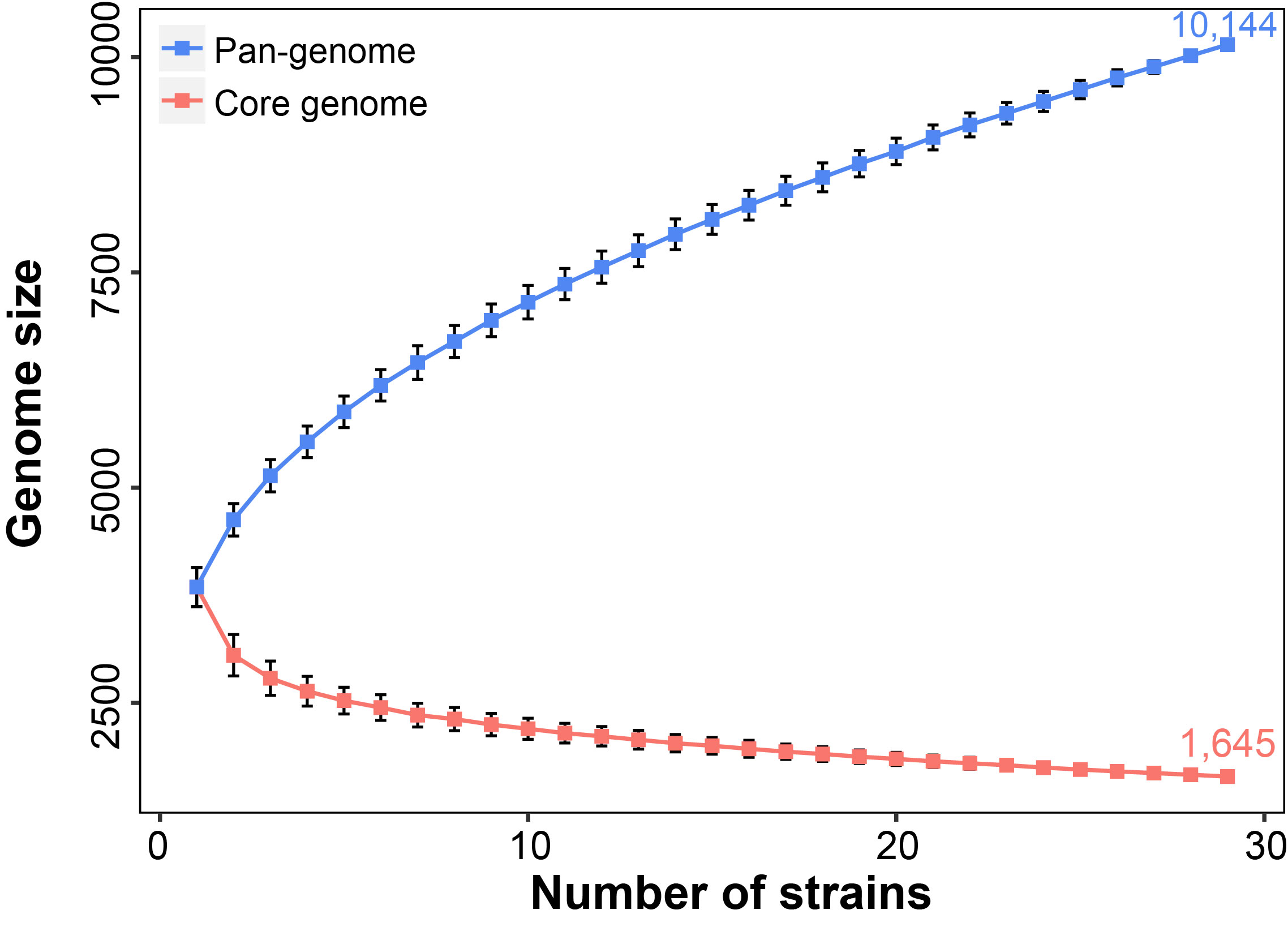
Pan-genome structure Aeromonas
The 29 Aeromonas species exhibited an open pan-genome structure, which tended to rise progressively. The increase of genomic data size would result in the expanding of pan-genome sizes and greater genomic diversity in Aeromonas genus. In contrast to the pan-genome, the estimated core-genome size of the 29 strains included in our analysis has gradually decreased and not reached a plateau. Both core and pan genome were still influenced by the inclusion of newly sequenced strains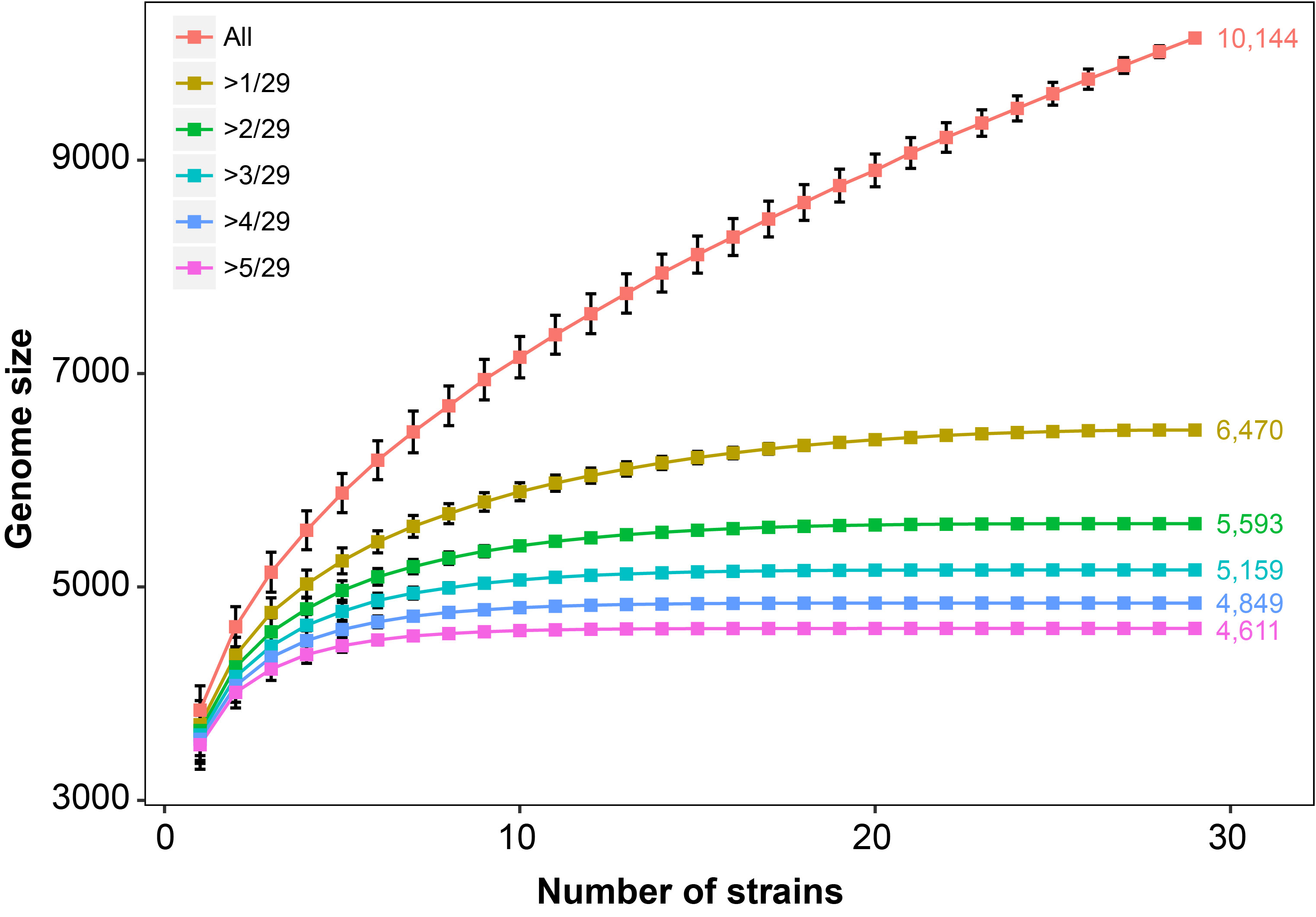 Rarefaction analysis of the number of non-redundant genes against the number of strains showed that
the genes shared by more than one strain (non-unique genes) were almost constant with approximately
29 strains, while genes unique for strains increased even beyond 29 strains
Rarefaction analysis of the number of non-redundant genes against the number of strains showed that
the genes shared by more than one strain (non-unique genes) were almost constant with approximately
29 strains, while genes unique for strains increased even beyond 29 strains
Functional characterization of the Aeromonas pan-genome
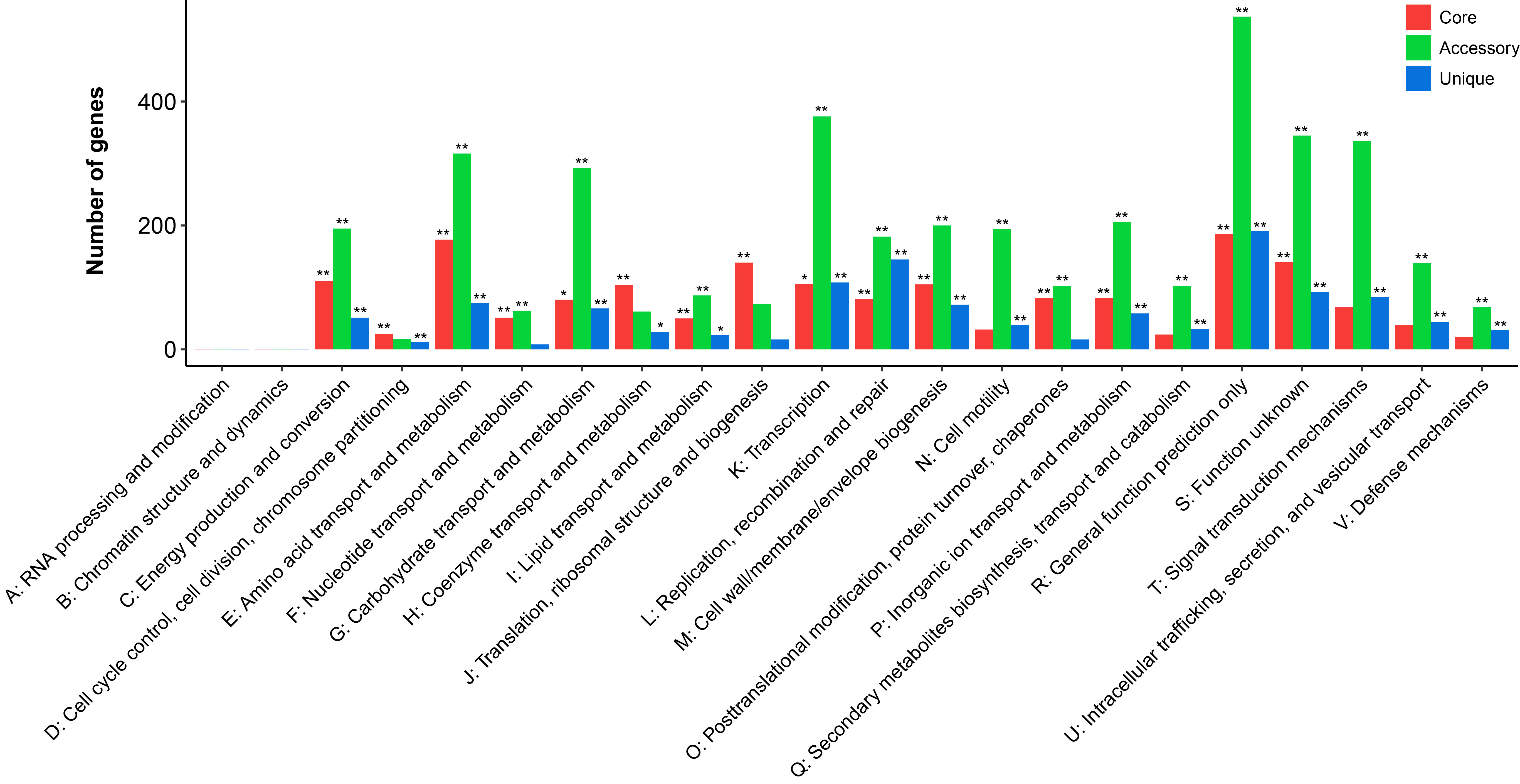
The core genome conferred an extensive functional repertoire involved in metabolism, which are required in maintaining basic cellular functions. Genes assigned to “transcription” (376 genes, Fisher's exact test p-value < 0.001), “amino acid transport and metabolism” (316 genes, Fisher's exact test p-value < 0.001), “carbohydrate transport and metabolism” (293 genes, Fisher's exact test p-value < 0.001), and “signal transduction mechanisms” (336 genes, Fisher's exact test p-value < 0.001) were prominently represented in the accessory component of the pan-genome. The number of genes encoding functions “cell cycle control, cell division, chromosome partitioning”, “coenzyme transport and metabolism” and “translation, ribosomal structure and biogenesis” were higher in the core genome than in the accessory and unique genome.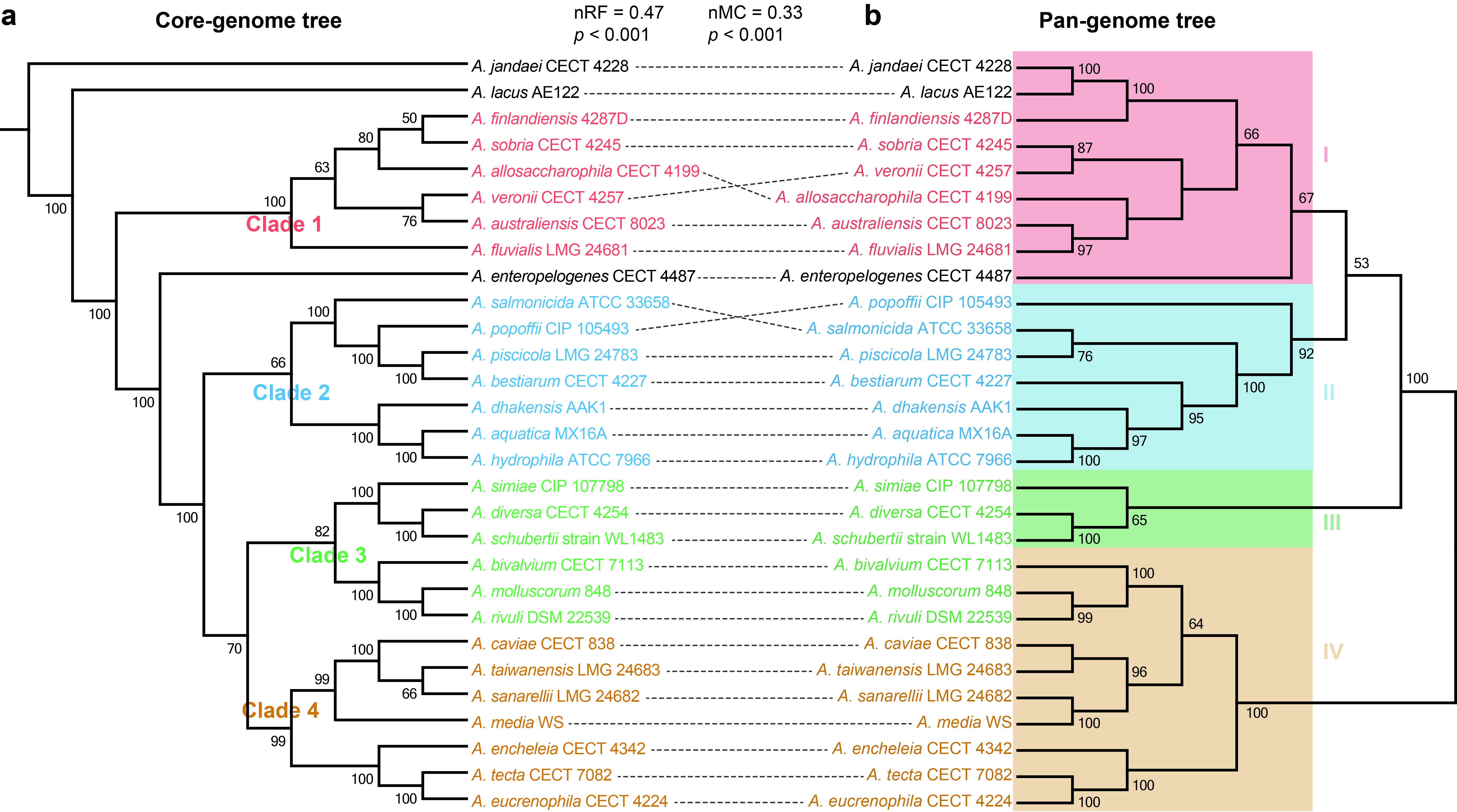
Phylogenetic relationships of Aeromonas species
Comparing phylogeny based on core genome to that based on pan-genome, we found that phylogenetic relationships among core genome bore a high resemblance with relationships among whole gene content (nRF = 0.47, t-test, p < 0.001; nMC = 0.33, t-test, p < 0.001), despite the occurrence of large number of variable genes. These results suggested that phylogenetic relationships among Aeromonas strains were mainly affected by the content of shared genes, the variable genes still accounted for an important proportion of phylogenetic signals, and genetic diversity was of great significance in evolution.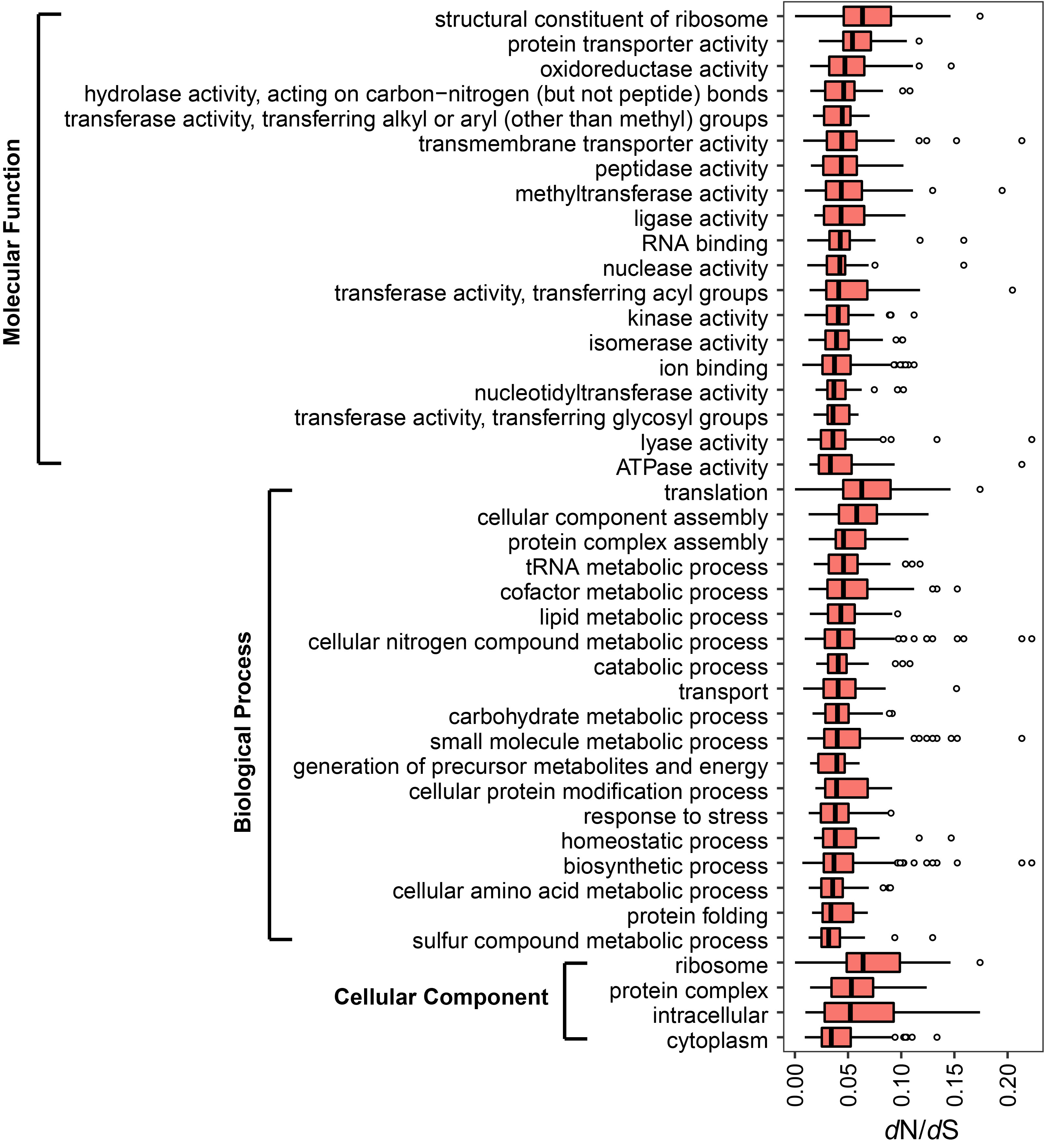
Selection constraints on gene functions in Aeromonas core and accessory genome
Selection constraints on gene functions in Aeromonas core and accessory genome In the core genome, genes involved in the “ATPase activity”, “sulfur compound metabolic process” and “cytoplasm” exhibited significant stronger evolutionary constraints than “structural constituent of ribosome”, “translation”, and “ribosome” in core genome (t-test, p < 0.001).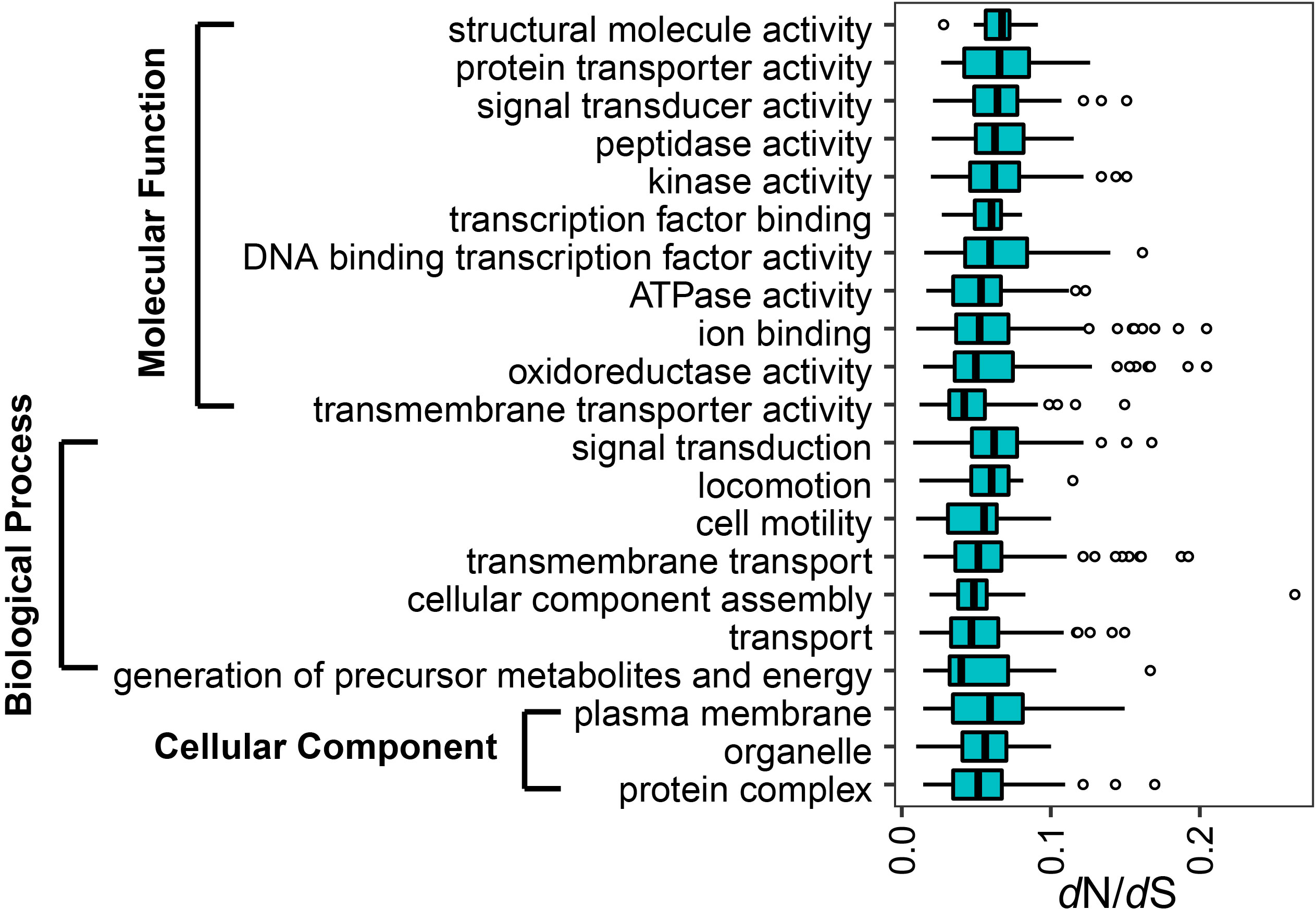
In the accessory genome, the “transmembrane transporter activity”, “generation of precursor metabolites and energy” and “protein complex” underwent relatively stronger constraints than other functions in molecular function, biological process and cellular component, respectively (t-test, p < 0.001).
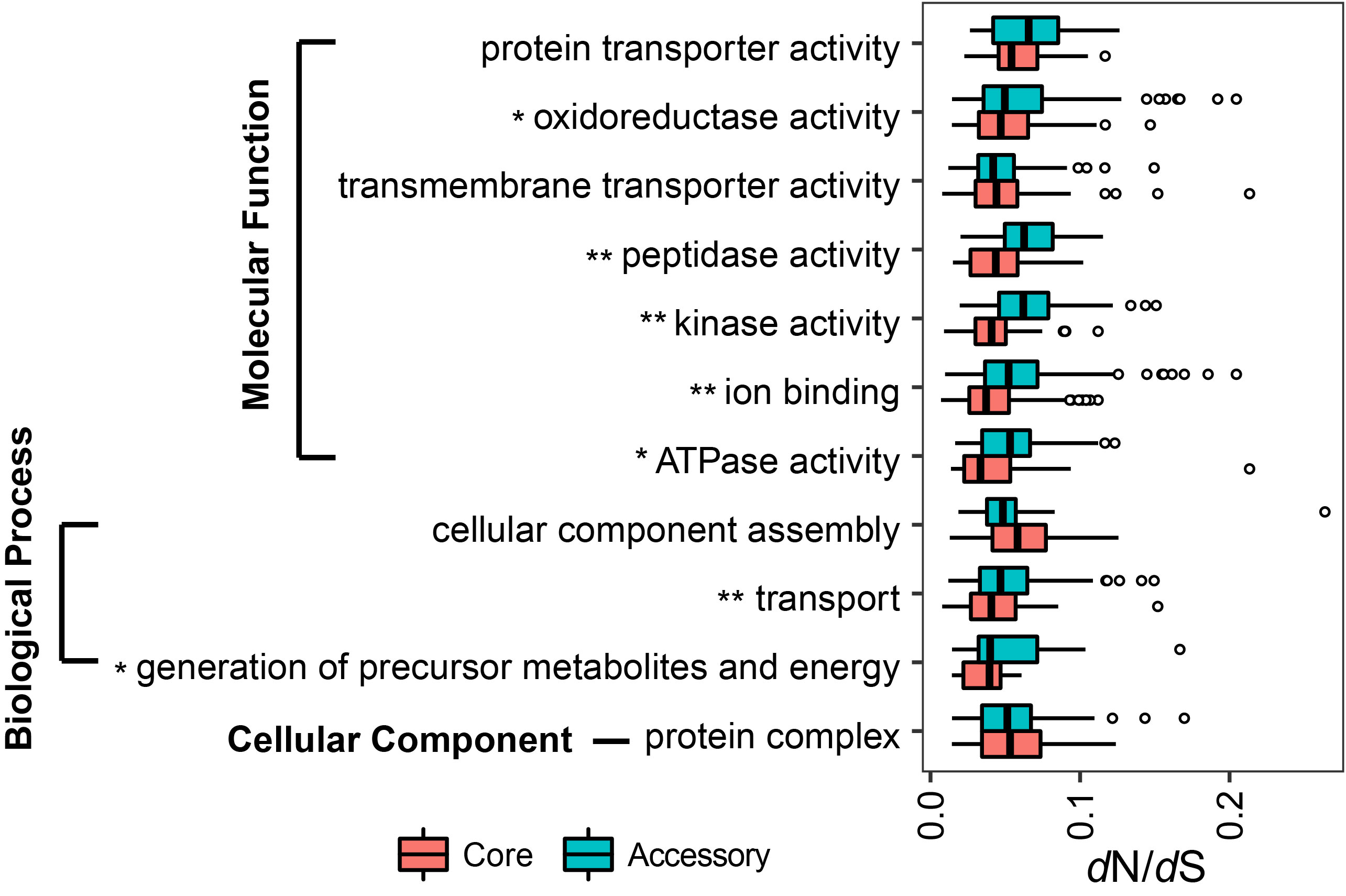
The core genes encoding the functions of “oxidoreductase activity”, “peptidase activity”, “kinase activity”, “ion binding”, “ATPase activity”, “transport” and “generation of precursor metabolites and energy” were under significantly stronger selective pressure than the corresponding accessory genes (t-test, p < 0.05).
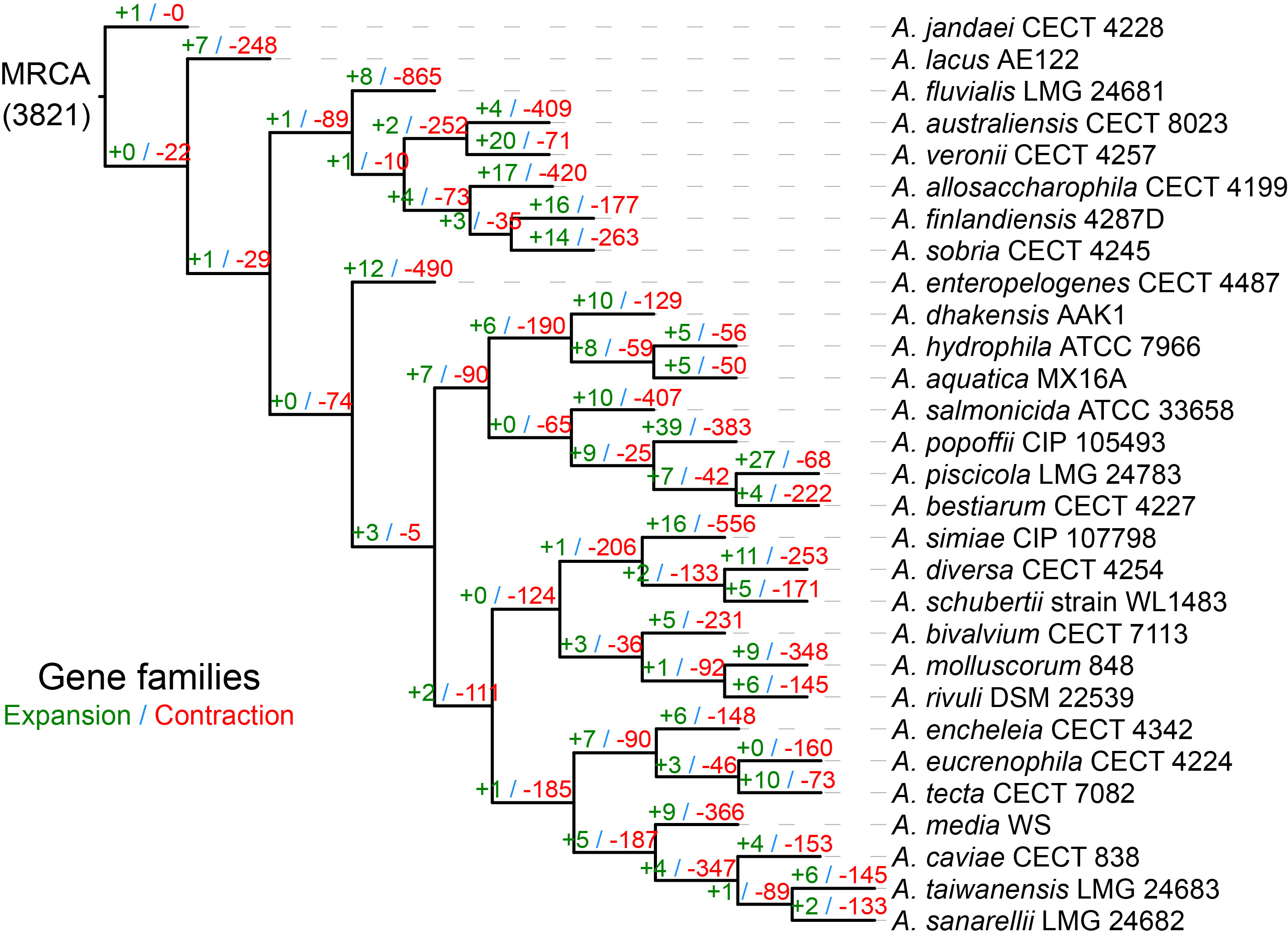
Gene family expansion and contraction of Aeromonas.
The evolutionary flexibility of these Aeromonas genomes was evident in the determinations of gene gain and loss on each of the respective lineages. Determinations of the number of genes expanded and contracted on each branch showed considerable variation, and the contraction was considerably greater than expansion, which was particularly evident on external branches.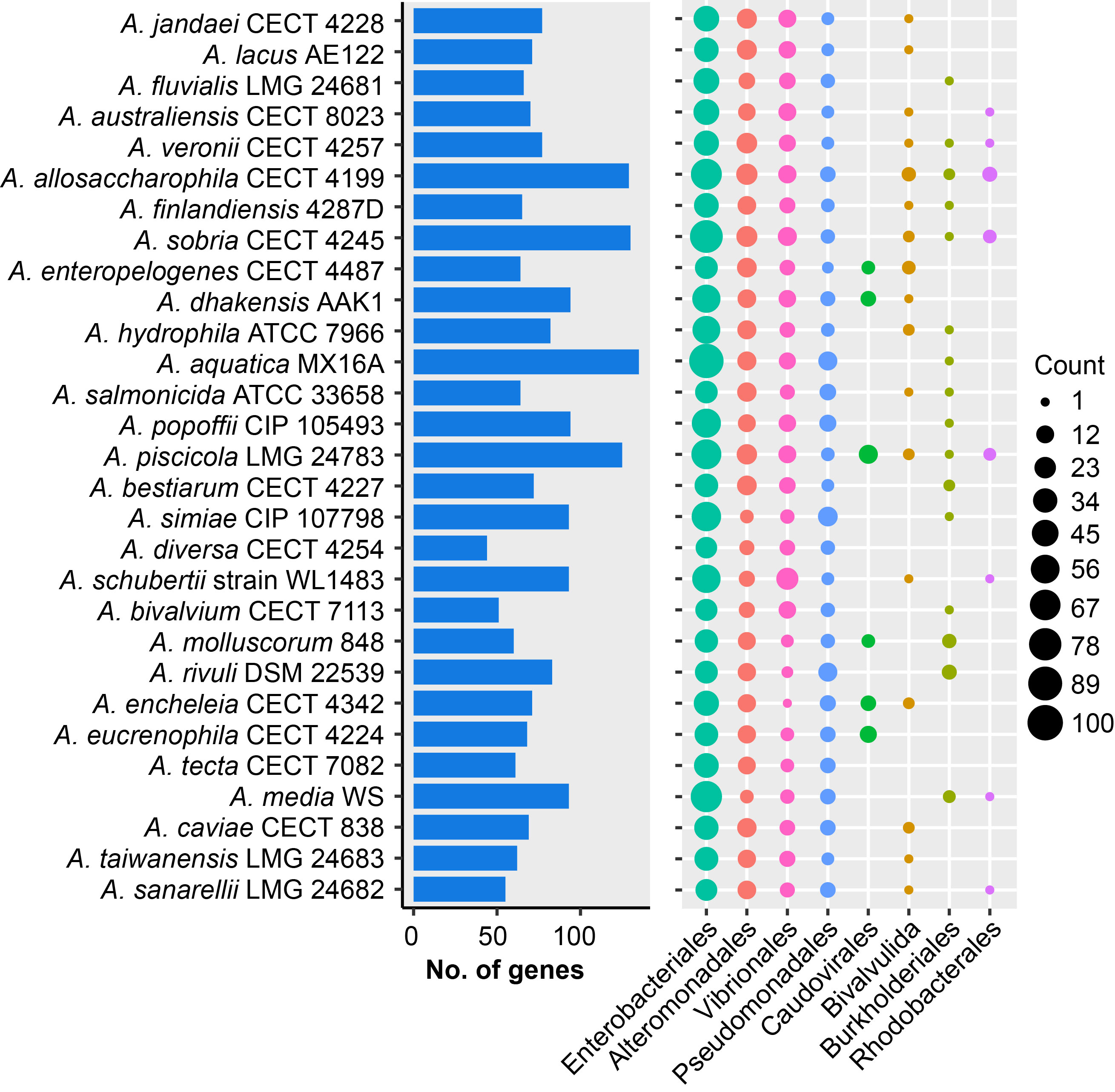
Distribution of horizontal genes acquired in each strain
We obtained 625 gene families that would be widespread via HGT in these strains, of which 249 were unique genes. These putatively transferred genes modulating gene inventory were mainly from Enterobacteriales, Alteromonadales and Vibrionales
Composition and distribution of mobile elements
we identified 421 gene families associated with MGEs in the pan-genome, of which 44.7% (188 out of 421) were strain specific. These MGEs were mainly composed of gene families associated with plasmids (232), with a minor fraction of gene families associated to IS (90), prophage (93) and viruses (6).
Distribution pattern of virulence factors in Aeromonas genomes
Aeromonas strains carrying different virulence genes may have an important impact on their pathogenicity and the presence of unique genes may be one of the factors on the ability of different pathogenicity. In addition, high levels of virulence gene gain and loss were evident in these bacterial genomes, which would enhance the evolutionary flexibility of these virulence genes.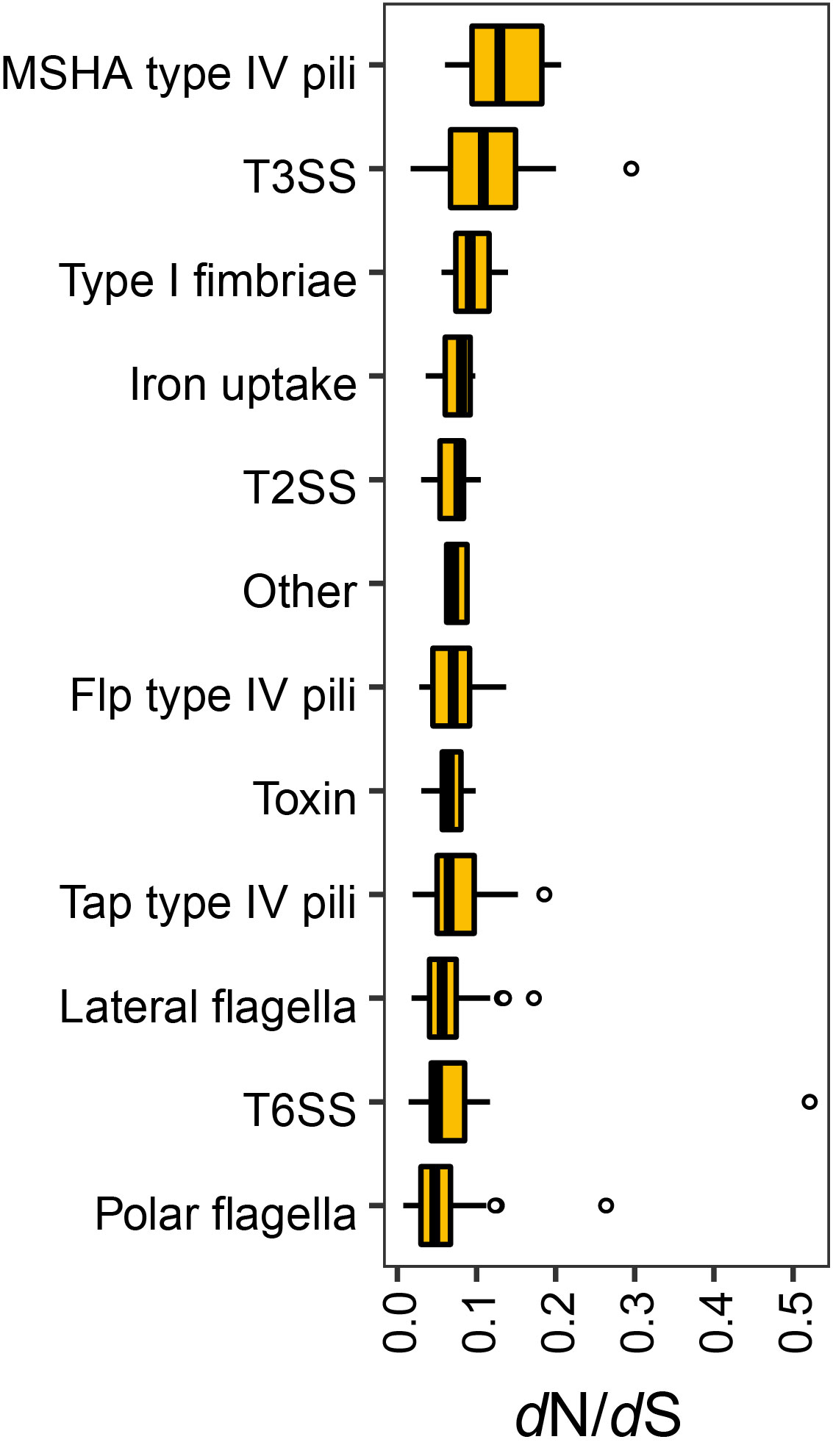 The single-copy virulence genes were subject to purifying selective pressure, which may be of major relevance for maintenance of pathogenicity.
The single-copy virulence genes were subject to purifying selective pressure, which may be of major relevance for maintenance of pathogenicity.
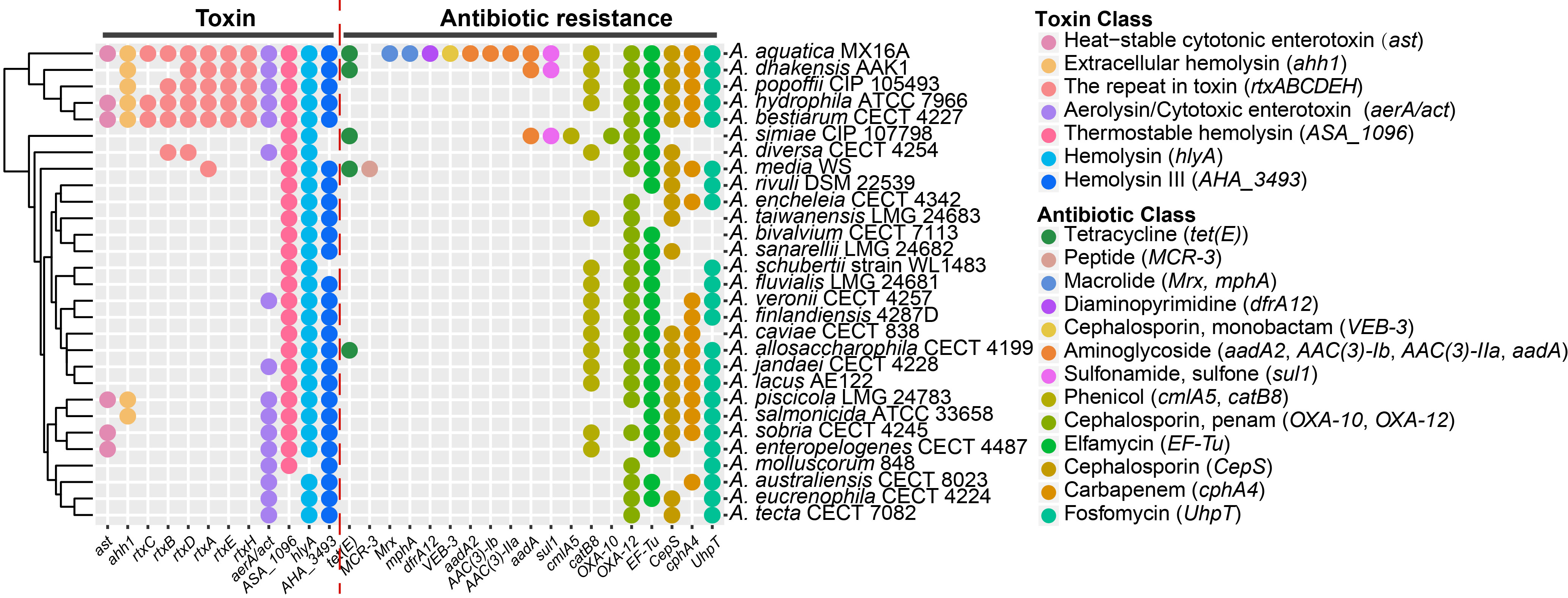
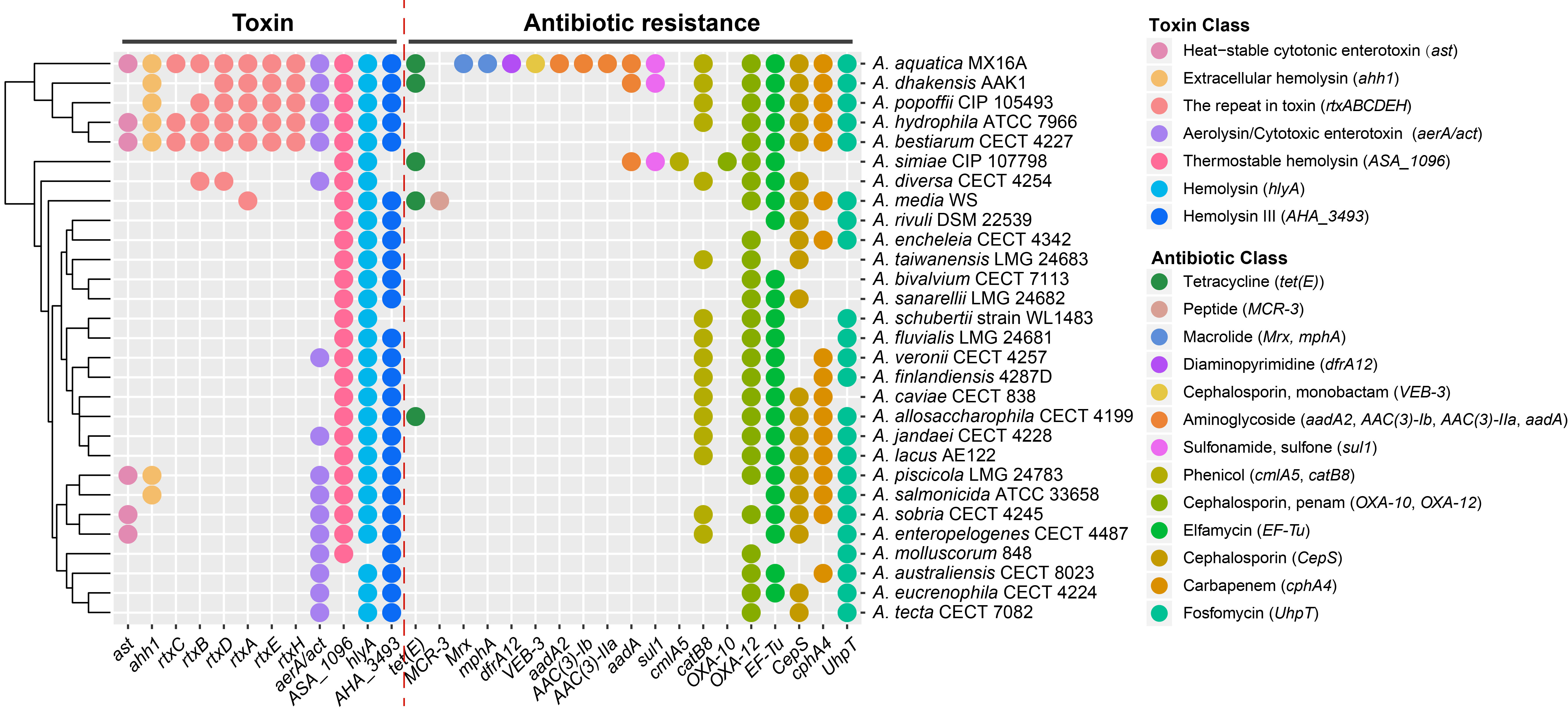 We observed the different distributions of the 12 genes encoding toxins across these genomes, which might contribute to the pathogenicity differences. The A. aquatica MX16A, A. dhakensis AAK1, A. popoffii CIP 105493, A. hydrophila ATCC 7966 and A. bestiarum CECT 4227 contained the most types of toxin coding genes. We also obtained 19 genes involved in a broad spectrum of antibiotic resistance, ranging from tetracycline antibiotic to fosfomycin antibiotic.
We observed the different distributions of the 12 genes encoding toxins across these genomes, which might contribute to the pathogenicity differences. The A. aquatica MX16A, A. dhakensis AAK1, A. popoffii CIP 105493, A. hydrophila ATCC 7966 and A. bestiarum CECT 4227 contained the most types of toxin coding genes. We also obtained 19 genes involved in a broad spectrum of antibiotic resistance, ranging from tetracycline antibiotic to fosfomycin antibiotic.
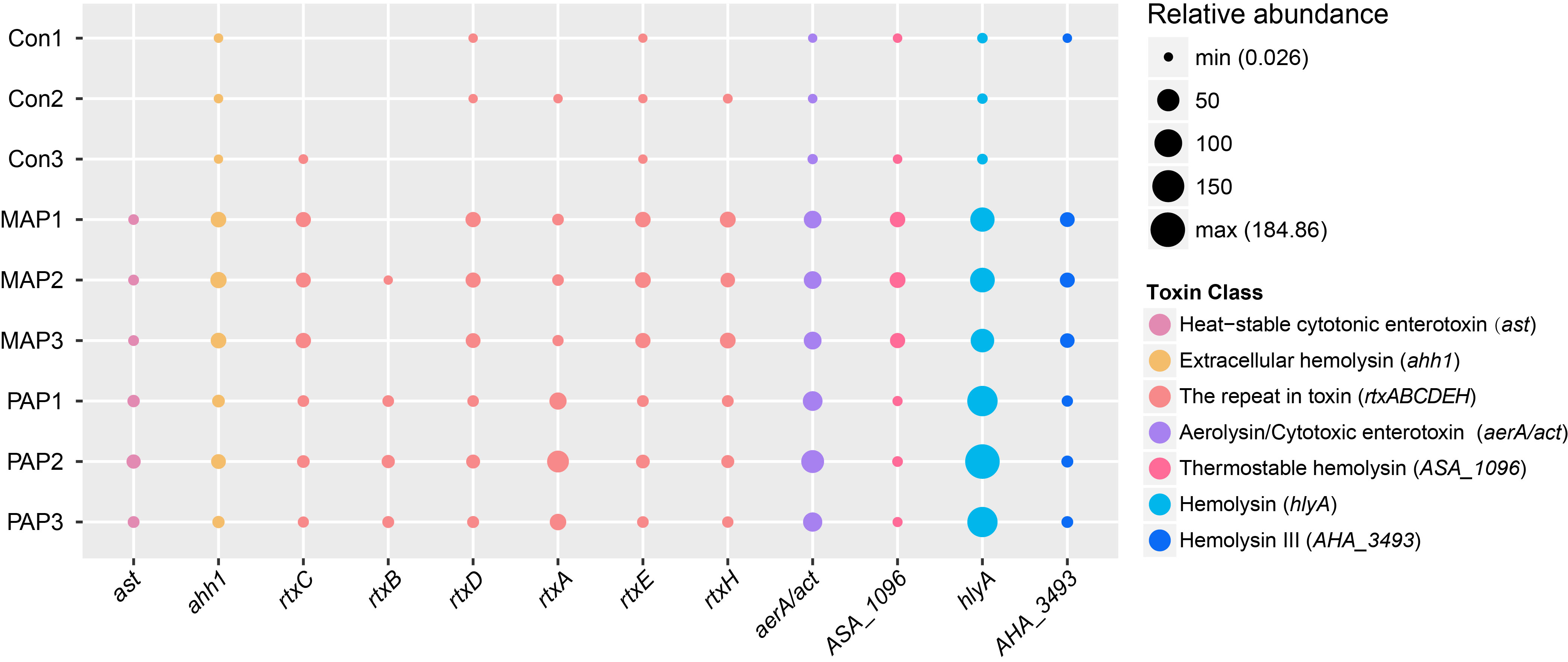 We also examined the different distributions of 12 toxin genes of Aeromonas in these samples, in which variation in toxin types would be the result of environmental changes. These results indicated that toxin genes of Aeromonas have differing patterns of co-occurrence over environment changes. The abundance of genes encoding hemolysin was higher than abundance of other toxins, which supported the universal retention of hemolysin pathogenicity in Aeromonas.
We also examined the different distributions of 12 toxin genes of Aeromonas in these samples, in which variation in toxin types would be the result of environmental changes. These results indicated that toxin genes of Aeromonas have differing patterns of co-occurrence over environment changes. The abundance of genes encoding hemolysin was higher than abundance of other toxins, which supported the universal retention of hemolysin pathogenicity in Aeromonas.
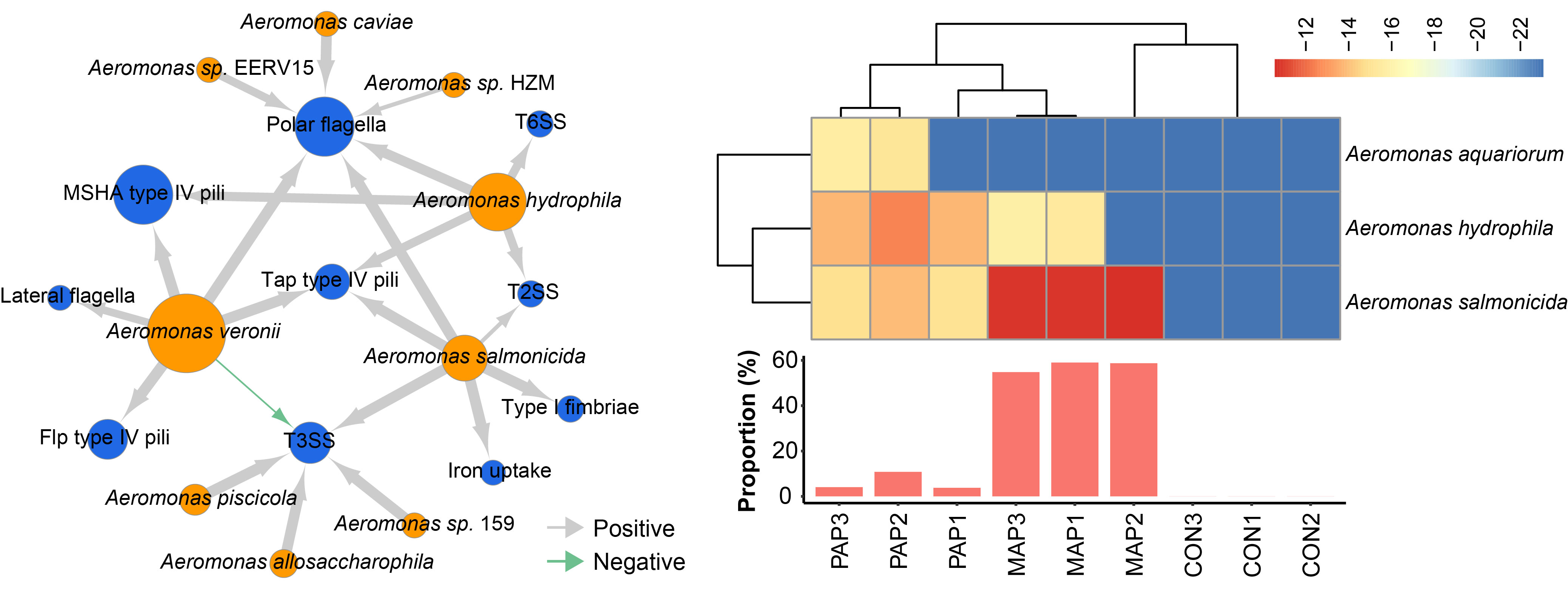
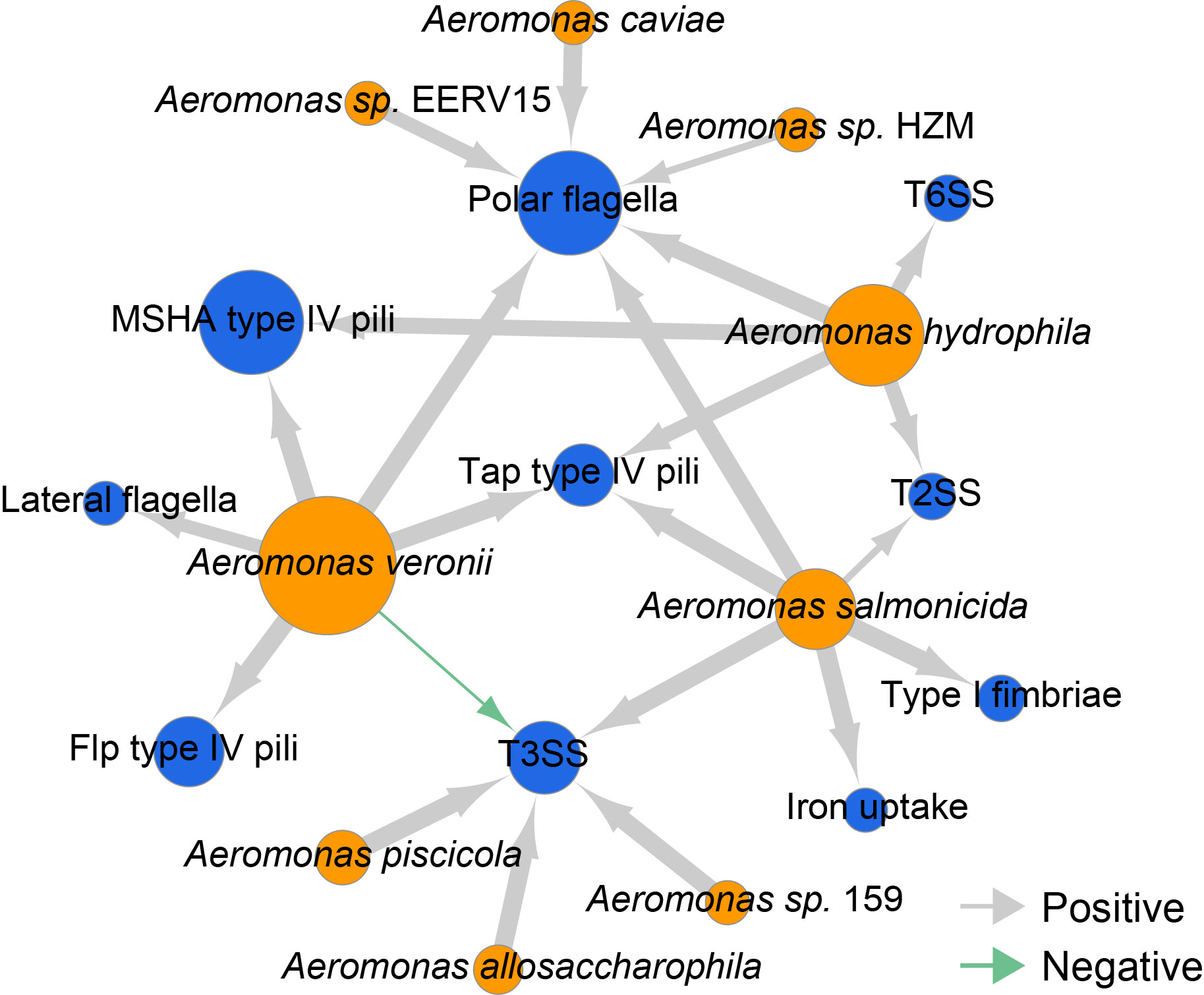 The co-occurrence of virulence genes and Aeromonas species showed that Aeromonas veronii, Aeromonas hydrophila and Aeromonas salmonicida carried genes involved in polar flagella and Tap type IV pili.
The co-occurrence of virulence genes and Aeromonas species showed that Aeromonas veronii, Aeromonas hydrophila and Aeromonas salmonicida carried genes involved in polar flagella and Tap type IV pili.
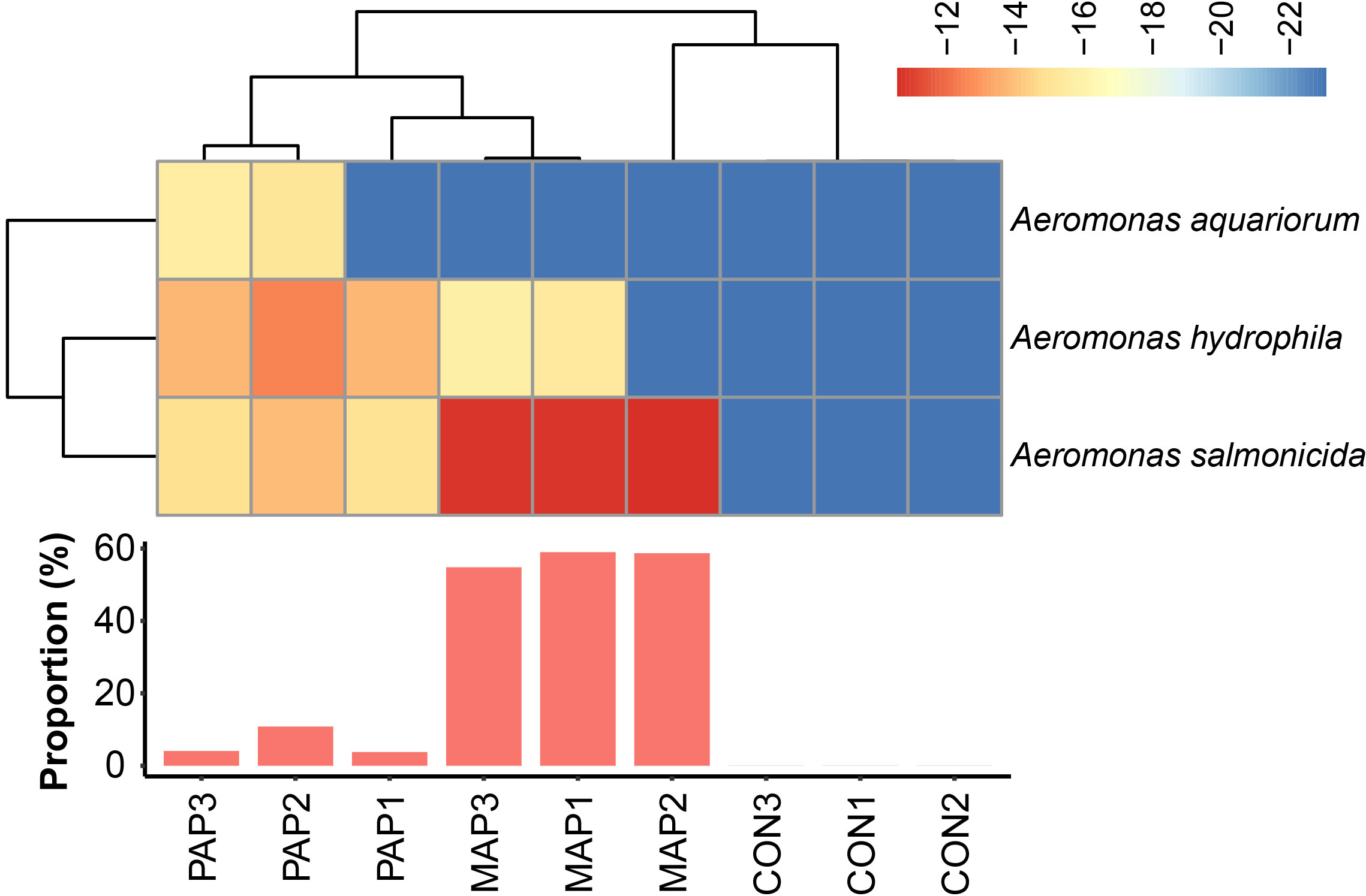 We detected a high abundance of polymyxin resistance pathways in these samples after different packaging, in which high abundance of polymyxin resistance from Aeromonas aquariorum, Aeromonas salmonicida and Aeromonas hydrophila and predominantly was originated from Aeromonas in MAP (57.5 ± 1.91%), indicating a possible acquisition of polymyxin resistance during environmental changes.
We detected a high abundance of polymyxin resistance pathways in these samples after different packaging, in which high abundance of polymyxin resistance from Aeromonas aquariorum, Aeromonas salmonicida and Aeromonas hydrophila and predominantly was originated from Aeromonas in MAP (57.5 ± 1.91%), indicating a possible acquisition of polymyxin resistance during environmental changes.